Genetik kalıtım, en basit anlatımda Mendel kurallarına göre işler: her birey, bir genin bir kopyasını anneden ve diğer kopyasını babadan alır ve her iki kopya da eşit derecede aktiftir. Bu genlerdeki özellikle baskın veya çekinik olmalarına ve, her iki kromozom üzerinde de aynı gen versiyonu olan allelin olmasına (homozigot) veya farklı allellerin olmasına (heterozigot) göre bireyin fenotipini belirler…
ANCAK, İNSAN MOLEKÜLER GENETİĞİ, ARTIK BUNUN ÇOK DAHA ÖTESİNE GEÇTİ…
Nitekim, insan genomunda bu kurala uymayan yüzlerce gen bulunmaktadır. Bu genler, ebeveynsel kökenlerine bağlı olarak aktif veya sessiz hale getirilirler. Bu olaya Genomik Damgalama (Genomic Imprinting) adı verilir ve bu hassas düzenlemedeki hatalar, nörogelişimsel bozuklukların en çarpıcı örneklerinden olan Angelman Sendromu (AS) ve Prader-Willi Sendromu (PWS)’na yol açar.
1. Genomik Damgalama (Genomic Imprinting) Mekanizması
Genomik damgalama, genlerin aktivasyonunun veya susturulmasının, bu genin anneden mi yoksa babadan mı kalıtıldığına bağlı olduğu bir epigenetik süreçtir.
- Epigenetik Kontrol: Damgalama, DNA dizisinde herhangi bir değişiklik yapmaz; bunun yerine, genin ekspresyonunu etkileyen kimyasal modifikasyonları (epigenetik işaretler) içerir.
- DNA Metilasyonu: En yaygın mekanizma, genin promotör bölgesindeki sitozin Nükleotid gruplarına bir metil grubu eklenmesidir. Genellikle, metilasyon genin susturulmasına (kapanmasına) neden olur.
- Histon Modifikasyonları: Kromatin yapısını gevşeterek veya sıkılaştırarak genin erişilebilirliğini ve dolayısıyla ekspresyonunu etkileyen modifikasyonlar da rol oynar.
Damgalama Kontrol Merkezi (IC/ICR): Bu epigenetik işaretler, her ebeveynin üreme hücrelerinde (sperm veya yumurta) kurulur ve yaşam boyu korunur. Bu işaretlerin kurulduğu spesifik DNA bölgesine Damgalama Kontrol Bölgesi (Imprinting Control Region – ICR) veya Damgalama Merkezi (Imprinting Center – IC) denir.
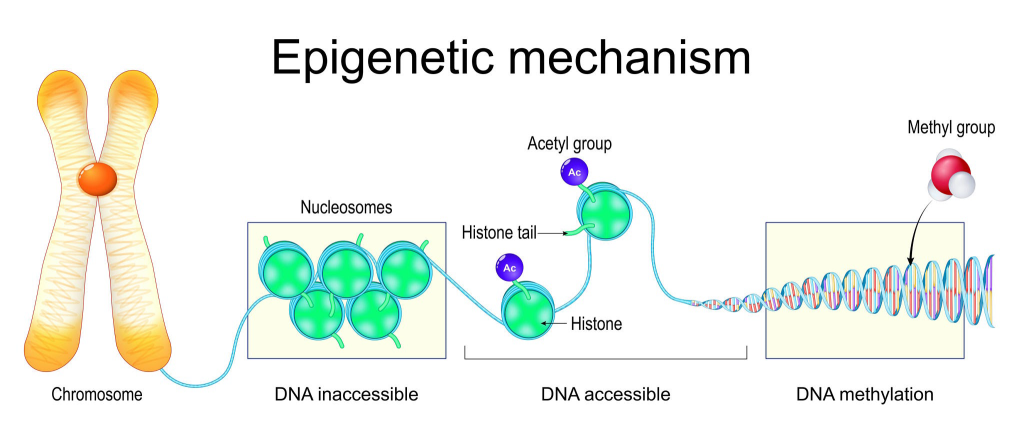
- Kalıtım: Çocuk, babadan metillenmiş (sessiz) bir kopya ve anneden metillenmemiş (aktif) bir kopya alabilir veya tam tersi. Bu ebeveyne özgü sessizleştirme, sağlıklı gelişim için zorunludur, çünkü bazı genlerin sadece bir kopyası (ya anneye ait ya da babaya ait olan) aktif olmalıdır.
2. Angelman ve Prader-Willi Sendromları: Bir Kromozom, İki Farklı Hastalık
AS ve PWS, genomik damgalamanın gücünü ve hassasiyetini gösteren iki zıt örnektir. Her iki sendrom da, Kromozom 15’in q11-q13 bölgesindeki damgalanmış genlerin düzgün çalışmamasından kaynaklanır [1, 2].
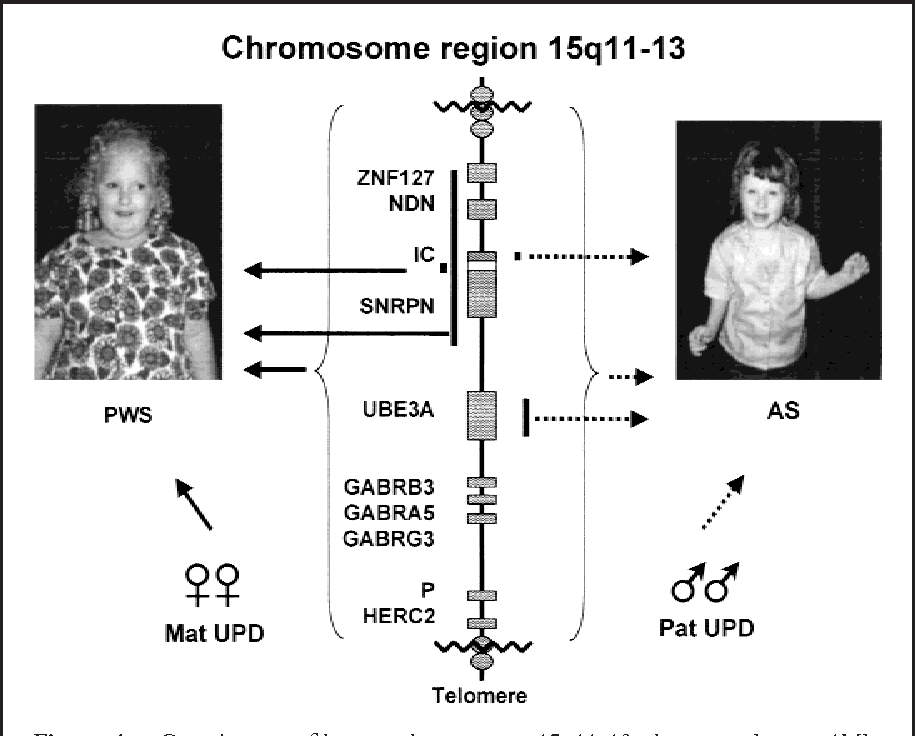
A. Angelman Sendromu (AS): Maternal Kopya Başarısızlığı
AS, esas olarak UBE3A genindeki fonksiyon kaybıyla ilişkilidir.
- Mekanizma: UBE3A geni, beyinde maternal olarak aktif bir gendir; yani normalde babadan gelen kopya sessiz (susturulmuş), anneden gelen kopya ise aktiftir [3].
- AS’li bireylerde, anneden gelen aktif kopya bozuktur veya yoktur (delinmiş/silinmiştir). Babadan gelen kopya zaten sessiz olduğu için, bireyin aktif ve çalışan hiçbir UBE3A kopyası kalmaz.
Genetik Nedenler:
- Maternal Bölge Silinmesi (%70): Kromozom 15’in maternal kopyasının silinmesi.
- Paternal Ünidisomi (%3-5): Birey, her iki kromozom 15’i de babadan almıştır. Babadan gelen kopyaların her ikisi de sessiz olduğundan, aktif UBE3A kopyası yoktur [2].
- IC Mutasyonları (%3): Damgalama Kontrol Bölgesi’ndeki (IC) mutasyonlar, maternal kopyanın yanlışlıkla sessizleştirilmesine neden olur.
- UBE3A Mutasyonu (%10): Genin kendi içindeki direkt nokta mutasyonları.
B. Prader-Willi Sendromu (PWS): Paternal Kopya Başarısızlığı
PWS, SNRPN ve NDN gibi kritik genlerin (bu genler normalde paternal olarak aktif genlerdir) fonksiyon kaybıyla ilişkilidir.
- Mekanizma: Bu genler, beyinde paternal olarak aktiftir; yani normalde anneden gelen kopya sessiz, babadan gelen kopya ise aktiftir [3].
- PWS’li bireylerde, babadan gelen aktif kopya bozuktur veya yoktur (silinmiş/delinmiştir). Anneden gelen kopya zaten sessiz olduğu için, bireyin aktif ve çalışan hiçbir kopyası kalmaz.
Genetik Nedenler:
- Paternal Bölge Silinmesi (%70): Kromozom 15’in paternal kopyasının silinmesi.
- Maternal Ünidisomi (%20-30): Birey, her iki kromozom 15’i de anneden almıştır. Anneden gelen kopyaların her ikisi de sessiz olduğundan, aktif paternal genlerin hiçbir kopyası kalmaz [2].
- IC Mutasyonları (%1-3): Damgalama Kontrol Bölgesi’ndeki mutasyonlar, paternal kopyanın yanlışlıkla sessizleştirilmesine neden olur.
3. Klinik Uygulama: Genetik Tanı ve Tedavi Stratejileri
Bu sendromların genetik kökenleri, kesin tanı ve tedavi stratejileri için kilit öneme sahiptir:
A. Tanı Yöntemleri
AS ve PWS’nin kesin tanısı için genetik testler kullanılır:
- DNA Metilasyon Testi: Bu, en güvenilir başlangıç testidir. Kromozom 15’in damgalanmış bölgesindeki metilasyon paternini analiz eder. PWS’de paternal aktif genler susturulmuş, AS’de maternal aktif genler susturulmuş gibi bir durum görülür. Bu test, silinme, ünidisomi ve IC mutasyonları dahil olmak üzere vakaların çoğunu teşhis edebilir [4].
- FISH (Floresan Yerinde Hibridizasyon) veya Kromozom Mikroarray: Silinmelerin tespitinde kullanılır.
- Mikrosatelit Analizi: Ünidisomi vakalarını (her iki kromozomun da aynı ebeveynden gelmesi) tespit etmek için kullanılır.
B. Tedavi ve Gelecek Perspektifleri
Güncel tedaviler semptom yönetimine odaklansa da, genetik damgalama mekanizmasının anlaşılması, yeni ve heyecan verici tedavi yollarının önünü açmıştır:
Yenilikçi tedavi Stratejileri: Her iki sendromda da sorun, bireyde susturulmuş (sessiz) ancak sağlam bir gen kopyasının bulunmasıdır (AS’de paternal kopya, PWS’de maternal kopya). Gen terapisi araştırmaları, sessiz olan kopyayı “uyandırmayı” veya damgalamayı tersine çevirmeyi hedeflemektedir. Örneğin, CRISPR/Cas9 teknolojisi ve nükleer antikorlar kullanılarak UBE3A’nın paternal kopyasındaki susturmayı kaldırarak AS’yi tedavi etme çalışmaları devam etmektedir [5].
Genomik damgalama mekanizmasının çözülmesi, bu nörogelişimsel bozuklukların sadece tanısını değil, gelecekteki epigenetik temelli küratif tedavilerini de mümkün kılacak bilimsel bir dönüm noktasıdır.
Yazar: Akın Sevinç
Editör: Umut Batuhan Sarı
Referanslar:
1. Cassidy, S. B., & Driscoll, D. J. (2009). Prader-Willi syndrome. Human Molecular Genetics.
2. Kishino, T., et al. (1997). The Angelman syndrome gene is imprinted in brain and absent in Prader-Willi syndrome. Nature Genetics.
3. Nicholls, R. D. (1994). Genomic imprinting and the Prader-Willi and Angelman syndromes. Annual Review of Genomics and Human Genetics.
4. Genetik Test Rehberleri. (2023). AS ve PWS için DNA metilasyon testlerinin ayırıcı tanıda kullanımı.
5. Gen Terapisi Çalışmaları. (2024). Targeting the paternal UBE3A allele for Angelman syndrome. Nature Biotechnology/Cell Press.
6. Unidisomi ve Delesyon Verileri. (Genetik Veritabanları)